4.1 Therapeutic indications
For the treatment of gastro-esophageal reflux disease (GERD) in patients who do not respond to PPI (proton-pump inhibitor) alone.
4.2 Posology and method of administration
1 capsule to be administered once daily. Zosa L Capsules should be administered on empty stomach, preferably in the morning or at least 1 hour prior to meal. The capsules should be swallowed whole with water and not to be opened, chewed or crushed. Or, as prescribed by the physician
Pediatric Patients
Safety and efficacy of esomeprazole with levosulpiride combination therapy has not been established in paediatric patients. Thus, Zosa L Capsules are not recommended for use in children and adolescents below 18 years of age.
Geriatric Patients
No dosage adjustment is generally necessary in the elderly patients with normal renal function, but dose should be reduced if there is evidence of renal impairment. Elderly patients are more susceptible to postural hypotension, sedation, and extrapyramidal side effects. Thus, caution should be exercised in the elderly population while on Zosa L therapy.
Renal Impairment Patients
Zosa L Capsules should be used with caution and dose/dosage frequency may need to be reduced depending on the severity of the renal dysfunction. Zosa L Capsules are contraindicated in patients with severe renal impairment.
Hepatic Impairment Patients
With esomeprazole, dose adjustment is not required in patients with mild to moderate liver impairment. For patients with severe liver impairment, a maximum dose of 20 mg esomeprazole should not be exceeded. There is no information available on use of levosulpiride in patients with hepatic dysfunction. Thus, as a precautionary measure, Zosa L Capsules should be avoided in patients with hepatic impairment.
4.3 Contraindications
- Patients with known hypersensitivity to esomeprazole or to any substituted benzimidazole derivative or to levosulpiride or to any component of the formulation.
- In patients receiving rilpivirine-containing products.
- Gastrointestinal bleeding and intestinal obstruction.
- Severe renal or hepatic insufficiency.
- Porphyrias.
- Alcohol intoxication.
- Certain tumors like phaeochromocytoma and pituitary prolactinoma.
- Concurrent use with levodopa or other antiparkinson drugs (including ropinirole).
4.4 Special warnings and precautions for use
Esomeprazole
Gastric Malignancy: In the presence of any alarm symptom (e.g., significant unintentional weight loss, recurrent vomiting, dysphagia, haematemesis or melaena) and when gastric ulcer is suspected or present, malignancy should be excluded, as treatment with esomeprazole may alleviate symptoms and delay diagnosis.
Helicobacter Pylori Eradication: When prescribing esomeprazole for eradication of Helicobacter pylori, possible drug interactions for all components in the triple therapy should be considered. Clarithromycin is a potent inhibitor of CYP3A4 and hence contraindications and interactions for clarithromycin should be considered when the triple therapy is used in patients concurrently taking other drugs metabolised via CYP3A4, such as cisapride.
Gastrointestinal Infections/Gastritis: Treatment with proton pump inhibitors (PPIs) may lead to a slightly increased risk of gastrointestinal infections such as Salmonella and Campylobacter. Atrophic gastritis has been noted occasionally in gastric corpus biopsies from patients treated long-term with omeprazole, of which esomeprazole is an enantiomer.
Clostridium Difficile-Associated Diarrhea (CDAD): Published observational studies suggest that PPI therapy like esomeprazole may be associated with an increased risk of Clostridium difficile-associated diarrhea, especially in hospitalized patients. This diagnosis should be considered for diarrhea that does not improve. Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated.
Absorption of Vitamin B12: Esomeprazole, like all acid-blocking medicines, may reduce the absorption of vitamin B12 (cyanocobalamin) due to hypo- or achlorhydria. This should be considered in patients with reduced body stores or risk factors for reduced vitamin B12 absorption on long-term therapy.
Risk of Bone Fracture: Several published observational studies suggest that PPI therapy may be associated with an increased risk for osteoporosis-related fractures of the hip, wrist, or spine. The risk of fracture was increased in patients who received high-dose (defined as multiple daily doses), and long-term PPI therapy (a year or longer). Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated. Patients at risk for osteoporosis-related fractures should be managed according to established treatment guidelines and they should have an adequate intake of vitamin D and calcium.
Subacute Cutaneous Lupus Erythematosus (SCLE): PPIs have been associated with cases of SCLE, although very infrequently. If lesions occur, especially in sun-exposed areas of the skin, and if accompanied by arthralgia, the patient should seek medical help promptly and esomeprazole therapy should be stopped immediately. The occurrence of SCLE with previous PPI treatment may increase the risk of SCLE with other PPIs.
Hypomagnesemia: Hypomagnesemia (symptomatic/asymptomatic), has been reported rarely in patients treated with PPIs for at least three months, in most cases after a year of therapy. Serious adverse events include tetany, arrhythmias, and seizures. In most patients, treatment of hypomagnesemia required magnesium replacement and discontinuation of the PPI. For patients expected to be on prolonged treatment or who take PPIs with medications such as digoxin or drugs that may cause hypomagnesemia (e.g., diuretics), it is recommended to monitor magnesium levels prior to initiation of PPI treatment, and periodically thereafter.
Levosulpiride
History of Breast Cancer: Levosulpiride may increase prolactin levels. Therefore, caution should be exercised and patients with a history or a family history of breast cancer should be closely monitored during levosulpiride therapy.
Prolongation of the QT Interval: Levosulpiride induces a prolongation of the QT interval. This effect is known to potentiate the risk of serious ventricular arrhythmias such as torsade de pointes. Levosulpiride should be used with caution in patients with cardiovascular disease or with a family history of QT prolongation.
Gastrointestinal Disorders: Levosulpiride should not be used when gastrointestinal stimulation of motility can be harmful e.g., in presence of gastrointestinal hemorrhage, mechanical obstructions or perforations.
Drugs Acting on CNS: Caution is advised when levosulpiride is administered concomitantly with other centrally acting drugs.
Alcohol: Concomitant intake of alcohol should be avoided during levosulpiride therapy as there is an increased chance of sedation.
Smoking: Smoking increases metabolism of the drug and thus, require higher dose of levosulpiride.
Parkinson’s Disease: In patient with Parkinson's disease, levosulpiride use should be avoided and an alternative drug therapy should be considered.
Convulsions: Cases of convulsions, sometimes in patients with no previous history, have been reported. In patients requiring levosulpiride who are receiving anticonvulsant therapy, the dose of the anticonvulsant should not be changed.
Anticholinergic Effects: Levosulpiride has an anticholinergic effect and, therefore, should be used with caution in patients with a history of glaucoma, ileus, congenital digestive stenosis, urine retention or hyperplasia of the prostate.
Hypertensive Patients: Levosulpiride should be used with caution in hypertensive patients, especially in the elderly population, due to the risk of hypertensive crisis. Patients should be adequately monitored.
4.5 Interaction with other medicinal products and other forms of interaction Esomeprazole
1) Interference with Antiretroviral Therapy Reduced Concentrations of Atazanavir and Nelfinavir: Concomitant use of atazanavir and nelfinavir with PPIs is not recommended. Co-administration of atazanavir with PPIs is expected to substantially decrease atazanavir plasma concentrations and may result in a loss of therapeutic effect and the development of drug resistance. If the combination of atazanavir with a PPI is unavoidable, close clinical monitoring is recommended in combination with an increase in the dose of atazanavir to 400 mg with 100 mg of ritonavir; esomeprazole 20 mg should not be exceeded.
Increased Concentrations of Saquinavir: Co-administration of saquinavir with PPIs is expected to increase saquinavir concentrations, which may increase toxicity and require dose reduction. Omeprazole, of which esomeprazole is an enantiomer, has been reported to interact with other antiretroviral drugs, too. The clinical importance and the mechanisms behind these interactions are not always known. Increased gastric pH during omeprazole treatment may change the absorption of the antiretroviral drug. Other possible interaction mechanisms are via CYP 2C19.
2) Drugs for Which Gastric PH Can Affect Bioavailability (ketoconazole, atazanavir, iron salts, erlotinib, mycophenolate mofetil, digoxin) Esomeprazole inhibits gastric acid secretion. Therefore, esomeprazole may interfere with the absorption of drugs where gastric pH is an important determinant of bioavailability. Like with other drugs that decrease intragastric acidity, the absorption of drugs such as ketoconazole, atazanavir, iron salts, erlotinib, and mycophenolate mofetil (MMF) can decrease, while the absorption of drugs such as digoxin can increase during treatment with esomeprazole. Digoxin: Concomitant treatment with omeprazole (20 mg daily), of which esomeprazole is an enantiomer, and digoxin in healthy subjects increased the bioavailability of digoxin by 10%. Co-administration of digoxin with esomeprazole is expected to increase the systemic exposure of digoxin. Therefore, patients may need to be monitored when digoxin is taken concomitantly with esomeprazole.
Mycophenolate Mofetil (MMF): Co-administration of omeprazole in healthy subjects and in transplant patients receiving MMF has been reported to reduce the exposure to the active metabolite, mycophenolic acid (MPA), possibly due to a decrease in MMF solubility at an increased gastric pH. The clinical relevance of reduced MPA exposure on organ rejection has not been established in transplant patients receiving esomeprazole and MMF. Use esomeprazole with caution in transplant patients receiving MMF.
3) Effects on Hepatic Metabolism/Cytochrome P-450 Pathways Esomeprazole is extensively metabolized in the liver by CYP 2C19 and CYP 3A4. In-vitro and in-vivo studies have shown that esomeprazole is not likely to inhibit CYPs 1A2, 2A6, 2C9, 2D6, 2E1, and 3A4. No clinically relevant interactions with drugs metabolized by these CYP enzymes would be expected. Drug interaction studies have shown that esomeprazole does not have any clinically significant interactions with quinidine, clarithromycin, or amoxicillin.
Warfarin: Patients treated with PPIs and warfarin concomitantly may need to be monitored for increases in INR (International Normalized Ratio) and prothrombin time. Increases in INR and prothrombin time may lead to abnormal bleeding.
Clopidogrel: Avoid concomitant use of esomeprazole with clopidogrel. Clopidogrel is a prodrug. Inhibition of platelet aggregation by clopidogrel is entirely due to an active metabolite. The metabolism of clopidogrel to its active metabolite can be impaired by use of concomitant medications, such as esomeprazole, that inhibit CYP2C19 activity. Concomitant use of clopidogrel with esomeprazole 40 mg reduces the pharmacological activity of clopidogrel. As a precaution, concomitant use of esomeprazole and clopidogrel should be discouraged or when using esomeprazole consider alternative anti-platelet therapy.
Diazepam: Esomeprazole may potentially interfere with CYP2C19, the major esomeprazole metabolizing enzyme. Co-administration of esomeprazole and diazepam, a CYP2C19 substrate, resulted in a 45% decrease in clearance of diazepam.
Phenytoin: Concomitant administration of esomeprazole 40 mg resulted in a 13% increase in trough plasma levels of phenytoin in epileptic patients. It is recommended to monitor the plasma concentrations of phenytoin when treatment with esomeprazole is introduced or withdrawn.
Cilostazol: Omeprazole as well as esomeprazole act as inhibitors of CYP2C19. Omeprazole, of which esomeprazole is an enantiomer, given in doses of 40 mg to healthy subjects in a cross-over study, increased Cmax and AUC for cilostazol by 18% and 26% respectively, and one of its active metabolites by 29% and 69%, respectively. Cisapride: In healthy volunteers, concomitant administration of esomeprazole 40 mg resulted in a 32% increase in area under the plasma concentration-time curve (AUC) and a 31% prolongation of elimination half-life (t½), but no significant increase in peak plasma levels of cisapride. The slightly prolonged QTc interval observed after administration of cisapride alone, was not further prolonged when cisapride was given in combination with esomeprazole. Voriconazole: Concomitant administration of esomeprazole and a combined inhibitor of CYP 2C19 and CYP3A4, such as voriconazole, may result in a more than doubling of the esomeprazole exposure. Dose adjustment of esomeprazole is not normally required. However, in patients with Zollinger-Ellison's Syndrome, who may require higher doses (up to 240 mg/day), dose adjustment may be considered. Rifampicin: Drugs known to induce CYP2C19 or CYP3A4 or both (such as rifampin) may lead to decreased esomeprazole serum levels. Avoid concomitant use of rifampin with esomeprazole. St. John's Wort: Omeprazole, of which esomeprazole is an enantiomer, has been reported to interact with St. John's wort, an inducer of CYP3A4. Avoid concomitant use of St. John's wort with esomeprazole.
4) Concomitant Administration with Other Drugs
Tacrolimus: Concomitant administration of esomeprazole and tacrolimus may increase the serum levels of tacrolimus.
Combination Therapy with Clarithromycin: Co-administration of esomeprazole, clarithromycin, and amoxicillin has resulted in an increase in plasma levels of esomeprazole and 14-hydroxyclarithromycin.
Methotrexate: Concomitant administration of PPIs and methotrexate (primarily at high dose) may elevate and prolong serum levels of methotrexate and/or its metabolite hydroxymethotrexate, leading to a risk of methotrexate toxicity. In high-dose methotrexate administration, a temporary withdrawal of the PPI may be considered in some patients.
5) Drug / Laboratory Tests Interactions
Interactions with Investigations of Neuroendocrine Tumors: Serum chromogranin A (CgA) levels increase secondary to drug-induced decreases in gastric acidity. The increased CgA level may cause false positive results in diagnostic investigations for neuroendocrine tumors. To avoid this interference, esomeprazole treatment should be stopped for at least 5 days before CgA measurements; consider repeating the test if initial CgA levels are high. Levosulpiride
Antacids and Sucralfate: Bioavailability of levosulpiride is reduced if it is taken concomitantly with sucralfate and aluminum/magnesium-containing antacids. So, these medicines should not be taken along with levosulpiride. There should be a minimum 2 hour time lag between the two medicines. Anticholinergic Drugs, Narcotics and
Analgesic Drugs: The effect of levosulpiride on gastrointestinal motility can be antagonized by these drugs. Antihypertensive Drugs: Concomitant use of levosulpiride may enhance the hypotensive effects of these drugs.
Anticholinergic Drugs: Concomitant administration may cause increase in incidence of anticholinergic side effects.
Levodopa/Antiparkinson Drugs (including ropinirole): There is reciprocal antagonism of effects between levodopa or antiparkinson drugs (including ropinirole) and levosulpiride. Levodopa reduces effects of levosulpiride; conversely, levosulpiride may decrease the efficacy of levodopa in the management of Parkinson’s disease. Thus, concomitant use of these drugs are contraindicated.
Atomoxetine, Antiarrhythmics, Terfenadine, Chloroquine, Quinine, Cisapride, and Drugs Causing Hypokalemia (corticosteroids, laxatives, and diuretics like furosemide): Concurrent use of levosulpiride with these drugs may cause arrhythmia, especially prolonged QT interval.
Alcohol: Levosulpiride can potentiate the cognitive and motor effects of alcohol. Thus, concurrent use should be avoided.
Lithium: Increased risk of extrapyramidal effects. Discontinuation of both drugs is recommended at first signs of neurotoxicity.
4.6 Use in special populations
Pregnancy Zosa L capsules are not recommended for use in pregnant women. Breast-feeding Zosa L capsules should not be used during breast feeding. Accordingly, a decision should be made whether to discontinue nursing or to discontinue/abstain from drug therapy, taking into account the benefit of the drug to the mother.
4.7 Effects on ability to drive and use machines
Patients should avoid driving or operating machinery or not to engage in activities that require full mental alertness.
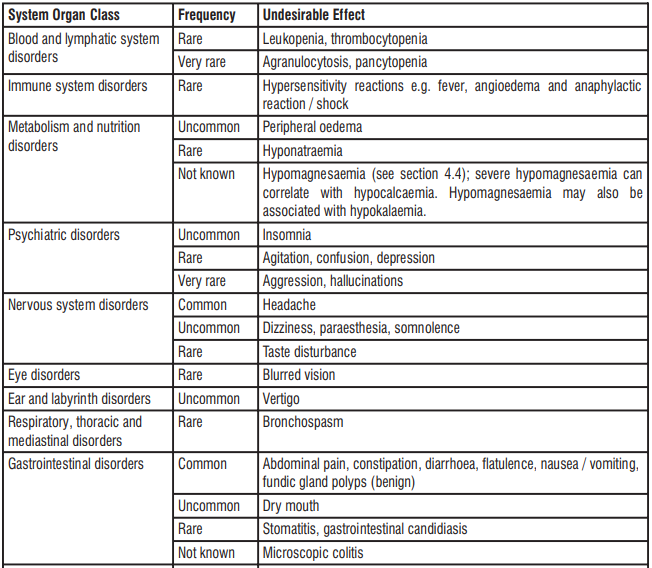
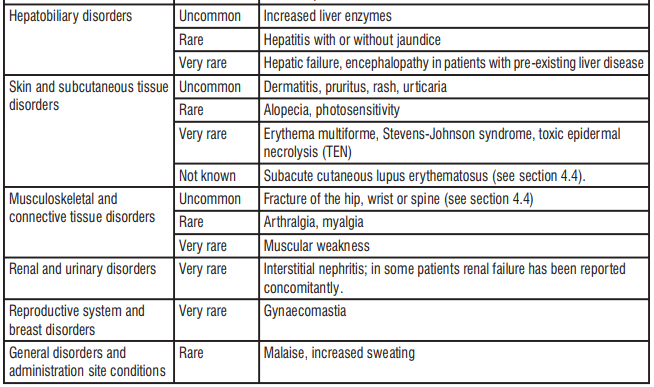
4.8 Undesirable effects
Esomeprazole
Acute kidney injury as an adverse drug reaction reported with the use of proton pump inhibitors.
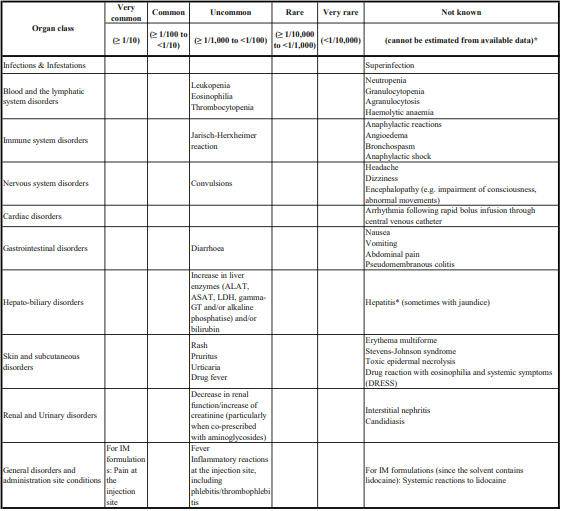
The most frequently reported (≥ 1%) adverse reactions with esomeprazole were headache, diarrhea, nausea, flatulence, abdominal pain, constipation, and dry mouth. Additional adverse reactions that were reported as possibly or probably related to esomeprazole with an incidence < 1% are as follows:
Body as a Whole: Enlarged abdomen, allergic reaction, asthenia, back pain, chest pain, substernal chest pain, facial edema, peripheral edema, hot flushes, fatigue, fever, flu-like symptoms, generalized edema, leg edema, malaise, pain, rigors.
Cardiovascular: Flushing, hypertension, tachycardia.
Endocrine: Goiter.
Gastrointestinal (GI): Bowel irregularity, constipation aggravated, dyspepsia, dysphagia, GI dysplasia, epigastric pain, eructation, esophageal disorders, frequent stools, gastroenteritis, GI hemorrhage, GI symptoms not otherwise specified, hiccup, melena, mouth disorders, pharyngeal disorders, rectal disorders, increase in serum gastrin, tongue disorders, tongue edema, ulcerative stomatitis, vomiting.
Hearing: Earache, tinnitus.
Hematologic: Anemia, cervical lymphadenopathy, epistaxis, leukocytosis, leukopenia, thrombocytopenia.
Hepatic: Abnormalities in hepatic function, including bilirubinemia, increase in AST (aspartate aminotransferase) and ALT (alanine aminotransferase).
Metabolic/Nutritional: Glycosuria, hyperuricemia, hyponatremia, increased alkaline phosphatase, thirst, vitamin B12 deficiency, weight increase/decrease.
Musculoskeletal: Arthropathy, cramps, fibromyalgia syndrome, hernia, polymyalgia rheumatica.
Nervous System/Psychiatric: Anorexia, apathy, increased appetite, confusion, depression aggravated, dizziness, hypertonia, nervousness, hypoesthesia, impotence, insomnia, migraine, paresthesia, sleep disorder, somnolence, tremor, vertigo, visual field defect.
Reproductive: Dysmenorrhea, menstrual disorders, vaginitis.
Respiratory: Aggravated asthma, coughing, dyspnea, laryngeal edema, pharyngitis, rhinitis, sinusitis.
Skin and Appendages: Acne, angioedema, dermatitis, pruritus, rash, urticaria, sweating.
Special Senses: Otitis media, parosmia, taste loss, taste perversion.
Urogenital: Albuminuria, cystitis, dysuria, fungal infection, hematuria, frequent micturition, moniliasis, polyuria.
Visual: Conjunctivitis, abnormal vision.
Laboratory Abnormalities
The following clinically significant laboratory changes in clinical trials, irrespective of relationship to esomeprazole, were reported in ≤ 1% of patients: Increased creatinine, uric acid, total bilirubin, alkaline phosphatase, ALT, AST, hemoglobin, white blood cell count, platelets, serum gastrin, potassium, sodium, thyroxine and thyroid stimulating hormone. Decreased levels of hemoglobin, white blood cell count, platelets, potassium, sodium, and thyroxine.
Post-Marketing Experience The following adverse reactions have been identified during post-marketing use of esomeprazole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic: Agranulocytosis, pancytopenia.
Eye: Blurred vision.
Gastrointestinal: Pancreatitis, stomatitis, microscopic colitis.
Hepatobiliary: Hepatitis with or without jaundice, hepatic failure.
Immune System: Anaphylactic reaction/shock.
Infections and Infestations: GI candidiasis, Clostridium difficile-associated diarrhea.
Metabolism and Nutritional Disorders: Hypomagnesemia.
Musculoskeletal and Connective Tissue: Muscular weakness, myalgia, fractures.
Nervous System: Hepatic encephalopathy, taste disturbance.
Psychiatric: Aggression, agitation, depression, hallucination.
Renal and Urinary: Interstitial nephritis.
Reproductive System and Breast: Gynecomastia.
Respiratory, Thoracic, and Mediastinal: Bronchospasm.
Skin and Subcutaneous Tissue: Alopecia, erythema multiforme, hyperhidrosis, photosensitivity, Stevens-Johnson syndrome, toxic epidermal necrolysis (sometime fatal), cutaneous lupus erythematosus.
Levosulpiride
The following side effects may occur with the use of levosulpiride: Acute muscular dystonia characterized by abnormal movements (twitching, tremor, etc.) of the hands, leg, tongue and facial muscles.
Sedation or drowsiness (because of decrease in sensory inputs to reticular activating system).
Increase in plasma prolactin levels manifested by breast enlargement (gynecomastia), production of milk (galactorrhea) and stopping of menstrual periods (amenorrhea).
Neuroleptic malignant syndrome (characterized by hyperpyrexia, muscle rigidity, increased myoglobin and creatine kinase).
Akathisia (uncontrollable desire to move about without any anxiety).
Tardive dyskinesia, it occurs late in the therapy and its features include involuntary rhythmical movements of face, mouth and jaw. The reason for tardive dyskinesia is synthesis of newer dopamine receptors which are supersensitive to even a small amount of dopamine. This causes a decrease in cholinergic activity in the striatum followed by decrease in gamma-amino butyric acid (GABA) release. This decreased in inhibitory GABA is responsible for increased involuntary motor activity.
Postural hypotension (because of autonomic blockade), tolerance develops to this effect after some time.
Weight gain. Elevated liver transaminases.
Reporting of suspected adverse reactions Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product.
Healthcare professionals are asked to report any suspected adverse reactions via
email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Experience with overdose is limited. There is no specific antidote. Treatment is only symptomatic. Appropriate supportive measures should therefore be instituted, close supervision of vital functions and cardiac monitoring (risk of QT interval prolongation and subsequent ventricular arrhythmias) is recommended until the patient recovers. If severe extrapyramidal symptoms occur anticholinergic drugs should be administrated. Overdose may be treated with alkaline osmotic diuresis and, if necessary, anti-parkinson drugs. Coma needs appropriate nursing, and cardiac monitoring is recommended until the patient recovers.