Zosa DSR Capsules


1.0 Generic Name
Esomeprazole (GR) & Domperidone (PR) Capsules
2.0 Qualitative and quantitative composition
Each hard gelatin capsule contains:
Esomeprazole Magnesium (Trihydrate) IP
equivalent to Esomeprazole …………………. 40 mg
(as gastro-resistant pellets)
Domperidone IP …………………………………… 30 mg
(as prolonged release pellets)
3.0 Dosage form and strength
Capsules, Esomeprazole (GR) & Domperidone (PR) [40 mg + 30 mg]
4.0 Clinical particulars
4.1 Therapeutic indication
For the treatment of adult patients with GERD (gastro esophageal reflux disease) not responding to esomeprazole alone.
4.2 Posology and method of administration
One capsule once daily.
Use in Special Populations
- Pediatric
Zosa DSR Capsules is not recommended in children below 12 years of age.
- Hepatic impairment
For patients with severe liver impairment, a maximum dose of 20 mg of esomeprazole should not be exceeded.
Domperidone is contraindicated in moderate or severe hepatic impairment.
Hence, Zosa DSR Capsules is contraindicated in patients with moderate or severe hepatic impairment.
- Renal impairment
Since the elimination half-life of domperidone is prolonged in severe renal impairment, Zosa DSR Capsules should be used with caution in patients with severe renal insufficiency. Patients on prolonged therapy should be reviewed regularly.
4.3 Contraindications
Zosa DSR Capsules is contraindicated in the following situations
- Known hypersensitivity to the active substance esomeprazole and domperidone, to substituted benzimidazoles or to any of the excipients listed in formulation.
- Prolactin-releasing pituitary tumour (prolactinoma)
- In patients who have known existing prolongation of cardiac conduction intervals, particularly QTc, patients with significant electrolyte disturbances or underlying cardiac diseases such as congestive heart failure.
This formulation should not be used when stimulation of gastric motility could be harmful: gastro-intestinal haemorrhage, mechanical obstruction or perforation.
4.4 Special warnings and precautions for use
- Symptomatic response to therapy with esomeprazole does not preclude the presence of gastric malignancy.
- Therapy with esomeprazole may be associated with Clostridium difficile associated diarrhea. Patients should use the lowest dose and shortest duration of PPI therapy appropriate.
- Atrophic Gastritis has been noted occasionally in gastric corpus biopsies from patients on long-term treatment with esomeprazole.
- For patients expected to be on prolonged esomeprazole treatment or who take PPIs with medications such as digoxin or drugs that may cause hypomagnesemia (e.g., diuretics), healthcare professionals may consider monitoring magnesium levels prior to initiation of PPI treatment and periodically thereafter.
- PPI therapy may be associated with an increased risk for osteoporosis-related fractures of the hip, wrist or spine. Patients should use the lowest dose and shortest duration of PPI therapy appropriate to the condition being treated.
- Serum chromogranin A (CgA) levels increase secondary to esomeprazole-induced decrease in gastric acidity. This may cause false-positive results in diagnostic investigations for neuroendocrine tumors. Providers should temporarily stop esomeprazole treatment before assessing CgA levels and consider repeating the test if initial CgA levels are high.
- Domperidone may be associated with an increased risk of serious ventricular arrhythmias or sudden cardiac death. Caution is exercised in patients who have existing prolongation of cardiac conduction intervals, electrolyte disturbances or underlying cardiac diseases.
- The risk of neurological side effects with domperidone is higher in young children. Overdosing may cause extra-pyramidal symptoms.
4.5 Drugs interactions
Esomeprazole
Effects of esomeprazole on the pharmacokinetics of other medicinal products
Protease inhibitors
Omeprazole has been reported to interact with some protease inhibitors. The clinical importance and the mechanisms behind these reported interactions are not always known. Increased gastric pH during omeprazole treatment may change the absorption of the protease inhibitors. Other possible interaction mechanisms are via inhibition of CYP 2C19.
For atazanavir and nelfinavir, decreased serum levels have been reported when given together with omeprazole and concomitant administration is not recommended. Co-administration of omeprazole (40 mg once daily) with atazanavir 300 mg/ritonavir 100 mg to healthy volunteers resulted in a substantial reduction in atazanavir exposure (approximately 75% decrease in AUC, Cmax and Cmin). Increasing the atazanavir dose to 400 mg did not compensate for the impact of omeprazole on atazanavir exposure. The co-administration of omeprazole (20 mg once daily) with atazanavir 400 mg/ritonavir 100 mg to healthy volunteers resulted in a decrease of approximately 30% in the atazanavir exposure as compared with the exposure observed with atazanavir 300 mg/ritonavir 100 mg once daily without omeprazole 20 mg once daily. Co-administration of omeprazole (40 mg once daily) reduced mean nelfinavir AUC, Cmax, and Cmin by 36–39 % and mean AUC, Cmax and Cmin for the pharmacologically active metabolite M8 was reduced by 75-92%. Due to the similar pharmacodynamic effects and pharmacokinetic properties of omeprazole and esomeprazole, concomitant administration with esomeprazole and atazanavir is not recommended and concomitant administration with esomeprazole and nelfinavir is contraindicated.
For saquinavir (with concomitant ritonavir), increased serum levels (80-100%) have been reported during concomitant omeprazole treatment (40 mg once daily). Treatment with omeprazole 20 mg once daily had no effect on the exposure of darunavir (with concomitant ritonavir) and amprenavir (with concomitant ritonavir). Treatment with esomeprazole 20 mg once daily had no effect on the exposure of amprenavir (with and without concomitant ritonavir). Treatment with omeprazole 40 mg once daily had no effect on the exposure of lopinavir (with concomitant ritonavir).
Methotrexate
When given together with PPIs, methotrexate levels have been reported to increase in some patients. In high-dose methotrexate administration a temporary withdrawal of esomeprazole may need to be considered.
Tacrolimus
Concomitant administration of esomeprazole has been reported to increase the serum levels of tacrolimus. A reinforced monitoring of tacrolimus concentrations as well as renal function (creatinine clearance) should be performed, and dosage of tacrolimus adjusted if needed.
Medicinal products with pH dependent absorption
Gastric acid suppression during treatment with esomeprazole and other PPIs might decrease or increase the absorption of medicinal products with a gastric pH dependent absorption. As with other medicinal products that decrease intragastric acidity, the absorption of medicinal products such as ketoconazole, atazanavir, iron salts, mycophenolate mofetil (MMF), itraconazole, and erlotinib can decrease and the absorption of digoxin can increase during treatment with esomeprazole. Concomitant treatment with omeprazole (20 mg daily) and digoxin in healthy subjects increased the bioavailability of digoxin by 10% (up to 30% in two out of ten subjects). Digoxin toxicity has been rarely reported. However, caution should be exercised when esomeprazole is given at high doses in elderly patients. Therapeutic medicinal product monitoring of digoxin should then be reinforced.
Co-administration of omeprazole in healthy subjects and in transplant patients receiving MMF has been reported to reduce the exposure to the active metabolite, mycophenolic acid (MPA), possibly due to a decrease in MMF solubility at an increased gastric pH. The clinical relevance of reduced MPA exposure on organ rejection has not been established in transplant patients receiving esomeprazole and MMF. Use esomeprazole with caution in transplant patients receiving MMF.
Medicinal products metabolised by CYP2C19
Esomeprazole inhibits CYP2C19, the major esomeprazole-metabolising enzyme. Thus, when esomeprazole is combined with medicinal products metabolised by CYP2C19, such as diazepam, citalopram, imipramine, clomipramine, phenytoin etc., the plasma concentrations of these medicinal products may be increased, and a dose reduction could be needed.
This should be considered especially when prescribing esomeprazole for on-demand therapy.
Diazepam
Concomitant oral administration of 30 mg esomeprazole resulted in a 45% decrease in clearance of the CYP2C19 substrate diazepam.
Phenytoin
Concomitant oral administration of 40 mg esomeprazole and phenytoin resulted in a 13% increase in trough plasma levels of phenytoin in epileptic patients. It is recommended to monitor the plasma concentrations of phenytoin when treatment with esomeprazole is introduced or withdrawn.
Voriconazole
Omeprazole (40 mg once daily) increased voriconazole (a CYP2C19 substrate) Cmax and AUCτ by 15% and 41%, respectively.
Cilostazol
Omeprazole as well as esomeprazole act as inhibitors of CYP2C19. Omeprazole, given in doses of 40 mg to healthy subjects in a cross-over study, increased Cmax and AUC for cilostazol by 18% and 26% respectively, and one of its active metabolites by 29% and 69% respectively.
Cisapride
In healthy volunteers, concomitant oral administration of 40 mg esomeprazole and cisapride resulted in a 32% increase in area under the plasma concentration-time curve (AUC) and a 31% prolongation of elimination half-life (t1/2) but no significant increase in peak plasma levels of cisapride. The slightly prolonged QTc interval observed after administration of cisapride alone, was not further prolonged when cisapride was given in combination with esomeprazole.
Warfarin
Concomitant oral administration of 40 mg esomeprazole to warfarin-treated patients in a clinical trial showed that coagulation times were within the accepted range. However, post-marketing of oral esomeprazole, a few isolated cases of elevated INR of clinical significance have been reported during concomitant treatment. Monitoring is recommended when initiating and ending concomitant esomeprazole treatment during treatment with warfarin or other coumarine derivatives.
Clopidogrel
Results from studies in healthy subjects have shown a pharmacokinetic (PK)/ pharmacodynamic (PD) interaction between clopidogrel (300 mg loading dose/75 mg daily maintenance dose) and esomeprazole (40 mg p.o. daily) resulting in decreased exposure to the active metabolite of clopidogrel by an average of 40% and resulting in decreased maximum inhibition of (ADP induced) platelet aggregation by an average of 14%.
When clopidogrel was given together with a fixed dose combination of esomeprazole 20 mg + ASA 81 mg compared to clopidogrel alone in a study in healthy subjects there was a decreased exposure by almost 40% of the active metabolite of clopidogrel. However, the maximum levels of inhibition of (ADP induced) platelet aggregation in these subjects were the same in the clopidogrel and the clopidogrel + the combined (esomeprazole + ASA) product groups.
Inconsistent data on the clinical implications of a PK/PD interaction of esomeprazole in terms of major cardiovascular events have been reported from both observational and clinical studies. As a precaution concomitant use of clopidogrel should be discouraged.
Investigated medicinal products with no clinically relevant interaction
Amoxicillin or quinidine
Esomeprazole has been shown to have no clinically relevant effects on the pharmacokinetics of amoxicillin or quinidine.
Naproxen or rofecoxib
Studies evaluating concomitant administration of esomeprazole and either naproxen or rofecoxib did not identify any clinically relevant pharmacokinetic interactions during short-term studies.
Effects of other medicinal products on the pharmacokinetics of esomeprazole
Medicinal products which inhibit CYP2C19 and/or CYP3A4
Esomeprazole is metabolised by CYP2C19 and CYP3A4. Concomitant oral administration of esomeprazole and a CYP3A4 inhibitor, clarithromycin (500 mg twice daily), resulted in a doubling of the exposure (AUC) to esomeprazole.
Concomitant administration of esomeprazole and a combined inhibitor of CYP2C19 and CYP 3A4 may result in more than doubling of the esomeprazole exposure. The CYP2C19 and CYP3A4 inhibitor voriconazole increased omeprazole AUCτ by 280%. A dose adjustment of esomeprazole is not regularly required in either of these situations. However, dose adjustment should be considered in patients with severe hepatic impairment and if long-term treatment is indicated.
Medicinal products which induce CYP2C19 and/or CYP3A4
Medicinal products known to induce CYP2C19 or CYP3A4 or both (such as rifampicin and St. John's wort) may lead to decreased esomeprazole serum levels by increasing the esomeprazole metabolism. Avoid concomitant use of St. John’s Wort or rifampin with esomeprazole.
Pediatric population
Interaction studies have only been performed in adults.
Domperidone
The main metabolic pathway of domperidone is through CYP3A4. In vitro data suggest that the concomitant use of drugs that significantly inhibit this enzyme may result in increased plasma levels of domperidone.
Increased risk of occurrence of QT-interval prolongation, due to pharmacodynamic and/or pharmacokinetic interactions.
Concomitant use of the following substances is contraindicated
QTc prolonging medicinal products
- anti-arrhythmics class IA (e.g., disopyramide, hydroquinidine, quinidine)
- anti-arrhythmics class III (e.g., amiodarone, dofetilide, dronedarone, ibutilide, sotalol)
- certain anti-psychotics (e.g., haloperidol, pimozide, sertindole)
- certain anti-depressants (e.g., citalopram, escitalopram)
- certain antibiotics (e.g. erythromycin, levofloxacin, moxifloxacin, spiramycin)
- certain antifungal agents (e.g., pentamidine)
- certain antimalarial agents (in particular halofantrine, lumefantrine)
- certain gastro-intestinal medicines (e.g., cisapride, dolasetron, prucalopride)
- certain antihistaminics (e.g., mequitazine, mizolastine)
- certain medicines used in cancer (e.g., toremifene, vandetanib, vincamine)
- certain other medicines (e.g., bepridil, diphemanil, methadone)
(see section Contraindications).
- apomorphine, unless the benefit of the co-administration outweighs the risks, and only if the recommended precautions for co-administration are strictly fulfilled.
Potent CYP3A4 inhibitors (regardless of their QT prolonging effects), i.e.:
- protease inhibitors
- systemic azole antifungals
- some macrolides (erythromycin, clarithromycin, telithromycin)
Concomitant use of the following substances is not recommended
Moderate CYP3A4 inhibitors i.e. diltiazem, verapamil and some macrolides.
Concomitant use of the following substances requires caution in use
Caution with bradycardia and hypokalaemia-inducing drugs, as well as with the following macrolides involved in QT-interval prolongation: azithromycin and roxithromycin (clarithromycin is contra-indicated as it is a potent CYP3A4 inhibitor).
The above list of substances is representative and not exhaustive.
Separate in vivo pharmacokinetic/pharmacodynamic interaction studies with oral ketoconazole or oral erythromycin in healthy subjects confirmed a marked inhibition of domperidone's CYP3A4 mediated first pass metabolism by these drugs.
With the combination of oral domperidone 10 mg four times daily and ketoconazole 200 mg twice daily, a mean QTc prolongation of 9.8 msec was seen over the observation period, with changes at individual time points ranging from 1.2 to 17.5 msec. With the combination of domperidone 10 mg four times daily and oral erythromycin 500 mg three times daily, mean QTc over the observation period was prolonged by 9.9 msec, with changes at individual time points ranging from 1.6 to 14.3 msec. Both the Cmax and AUC of domperidone at steady state were increased approximately three-fold in each of these interaction studies. In these studies domperidone monotherapy at 10 mg given orally four times daily resulted in increases in mean QTc of 1.6 msec (ketoconazole study) and 2.5 msec (erythromycin study), while ketoconazole monotherapy (200 mg twice daily) led to increases in QTc of 3.8 and 4.9 msec, respectively, over the observation period.
4.6 Use in special populations
Pregnancy
There are no adequate and well-controlled studies in pregnant women. Zosa DSR capsules should be used during pregnancy only if clearly needed.
Lactation
Domperidone is excreted in human milk and breast-fed infants receive less than 0.1% of the maternal weight-adjusted dose. Caution should be exercised when Zosa DSR Capsules is administered to a nursing mother.
4.7 Effects on ability to drive and use machines
Zosa DSR has minor influence on the ability to drive and use machines. Adverse reactions such as dizziness (uncommon) and blurred vision (rare) have been reported. If affected patients should not drive or use machines.
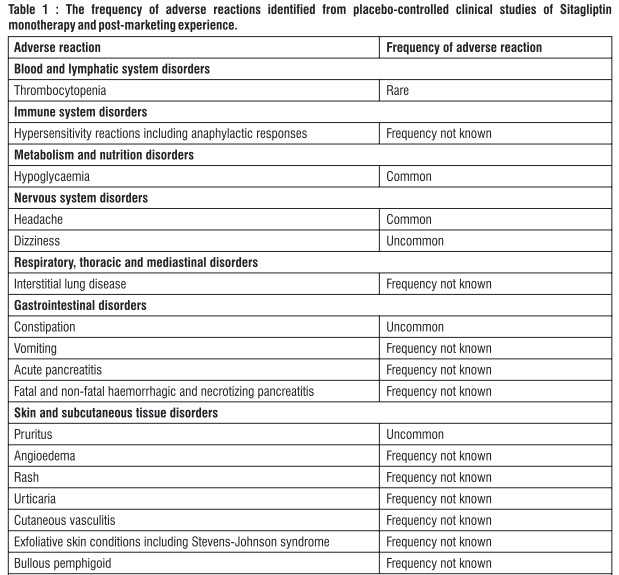
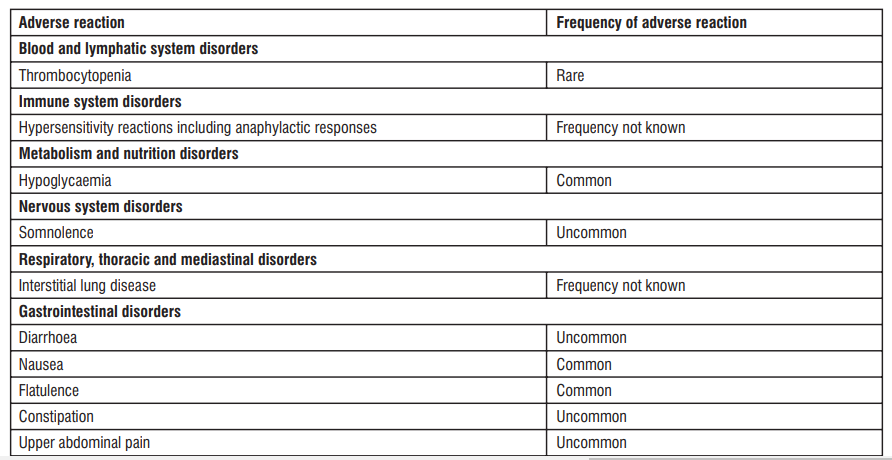
4.8 Undesirable effects
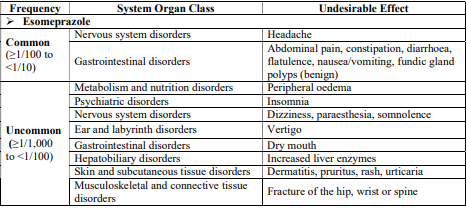
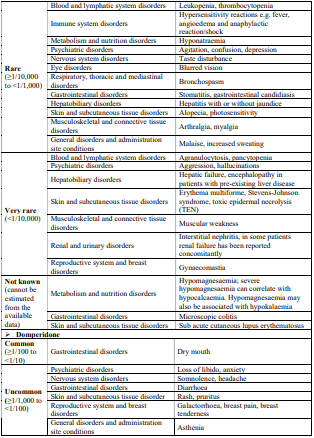
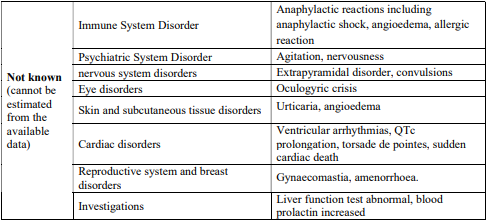
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via email to: medico@zuventus.com
Website: https://www.zuventus.com/drug-safety-reporting
By reporting side effects, you can help provide more information on the safety of this medicine.
4.9 Overdose
Overdose with 280 mg of esomeprazole were associated with gastrointestinal symptoms and weakness. Symptoms with domperidone over dosage may include agitation, altered consciousness, convulsions, disorientation, somnolence and extra pyramidal reactions. There is no specific antidote to esomprazole and domperidone.
In case of overdose, standard symptomatic treatment should be given immediately. Gastric lavage as well as the administration of activated charcoal may be useful. ECG monitoring should be undertaken. Close medical supervision and supportive therapy is recommended. Anticholinergic, anti-parkinson drugs may be helpful in controlling the extra pyramidal reactions seen with domperidone.
5.0 Pharmacological properties
5.1 Mechanism of action
Esomeprazole is a weak base and is concentrated and converted to the active form in the highly acidic environment of the secretory canaliculi of the parietal cell, where it inhibits the enzyme H+K + -ATPase – the acid pump and inhibits both basal and stimulated acid secretion.
Domperidone binds to dopamine receptor D2 (D2R) expressed by peripheral neurons; this inhibits dopamine binding and D2R-mediated signaling. Inhibition of peripheral D2R signaling prevents or relieves various gastrointestinal (GI) symptoms, such as nausea and vomiting, and may help relief reflux and symptoms of a variety of other upper GI disorders.
5.2 Pharmacodynamic properties
Esomeprazole
After 5 days of oral dosing with esomeprazole, intragastric pH above 4 was maintained for a mean time of 13 hours and 17 hours, respectively over 24 hours in symptomatic GERD patients. The proportion of patients maintaining an intragastric pH above 4 for at least 8, 12 and 16 hours respectively was 97%, 92% and 56%. Healing of reflux oesophagitis with esomeprazole occurs in approximately 78% of patients after 4 weeks, and in 93% after 8 weeks. During treatment with antisecretory medicinal products, serum gastrin and Chromogranin A (CgA) increases in response to the decreased acid secretion. Gastric glandular cysts have been reported to occur at a some what increased frequency on long-term treatment. These changes are benign and appear to be reversible.
Domperidone
Domperidone does not readily cross the blood brain barrier. In domperidone users, especially in adults, extra pyramidal side effects are very rare, but domperidone promotes the release of prolactin from the pituitary. Studies have shown oral domperidone to increase lower oesophageal pressure, improve antroduodenal motility and accelerate gastric emptying. There is no effect on gastric secretion.
When domperidone was administered in healthy subjects at up to 80 mg/day (i.e., more than twice the maximum recommended dosing) in QT study performed in accordance with ICH-E14 guidelines, no clinically relevant QTc effects were observed.
5.3 Pharmacokinetic properties
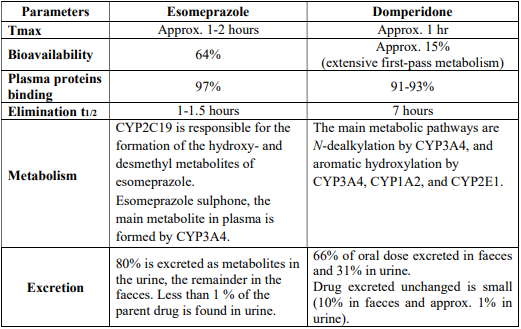
6.0 Nonclinical properties
6.1 Animal Toxicology or Pharmacology
Esomeprazole
Carcinogenicity: The carcinogenic potential of esomeprazole magnesium was assessed using studies of omeprazole, of which esomeprazole is an enantiomer. In two 24-month oral carcinogenicity studies in rats, omeprazole at daily doses of 1.7 mg/kg/day, 3.4 mg/kg/day, 13.8 mg/kg/day, 44 mg/kg/day, and 140.8 mg/kg/day (about 0.4 to 34 times the human dose of 40 mg/day expressed on a body surface area basis) produced gastric ECL cell carcinoids in a dose-related manner in both male and female rats; the incidence of this effect was markedly higher in female rats, which had higher blood levels of omeprazole.
In addition, ECL cell hyperplasia was present in all treated groups of both sexes. In one of these studies, female rats were treated with 13.8 mg omeprazole/kg/day (about 3.4 times the human dose of 40 mg/day on a body surface area basis) for 1 year, then followed for an additional year without the drug. No carcinoids were seen in these rats. An increased incidence of treatment-related ECL cell hyperplasia was observed at the end of 1 year (94% treated vs. 10% controls). By the second year the difference between treated and control rats was much smaller (46% vs. 26%) but still showed more hyperplasia in the treated group.
Mutagenesis: Esomeprazole was negative in the Ames mutation test, in the in vivo rat bone marrow cell chromosome aberration test, and the in vivo mouse micronucleus test.
Esomeprazole, however, was positive in the in vitro human lymphocyte chromosome aberration test.
Impairment of Fertility: The potential effects of esomeprazole on fertility and reproductive performance were assessed using omeprazole studies. Omeprazole at oral doses up to 138 mg/kg/day in rats (about 34 times the human dose of 40 mg/day on a body surface area basis) was found to have no effect on reproductive performance of parental animals.
Reproduction Studies: Reproduction studies have been performed in rats at oral doses up to 280 mg/kg/day (about 68 times an oral human dose of 40 mg on a body surface area basis) and in rabbits at oral doses up to 86 mg/kg/day (about 42 times an oral human dose of 40 mg on a body surface area basis) and have revealed no evidence of impaired fertility or harm to the fetus due to esomeprazole.
Domperidone
Safety margins in in vitro proarrhythmic models (isolated Langendorff perfused heart) exceeded the free plasma concentrations in humans at maximum daily dose (10 mg administered 3 times a day) by 9- up to 45-fold. In in vivo models the no effect levels for QTc prolongation in dogs and induction of arrhythmias in a rabbit model sensitized for torsade de pointes exceeded the free plasma concentrations in humans at maximum daily dose (10 mg administered 3 times a day) by more than 22-fold and 435-fold, respectively. In the anesthetized guinea pig model following slow intravenous infusions, there were no effects on QTc at total plasma concentrations of 45.4ng/ml, which are 3-fold higher than the total plasma levels in humans at maximum daily dose (10 mg administered 3 times a day).
At a high, maternally toxic dose (more than 40 times the recommended human dose), teratogenic effects were seen in the rat. No teratogenicity was observed in mice and rabbits. Development abnormalities observed in rats at a high exposure. Risk of carcinogenicity, mutagenicity or sensitization cannot be excluded.
7.0 Description
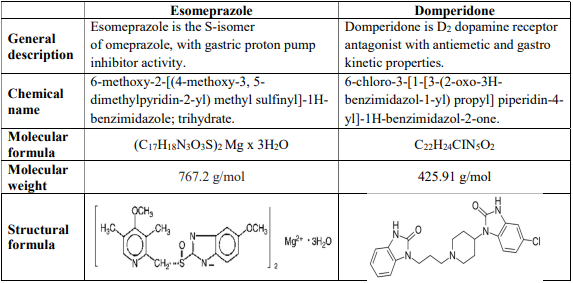
8.0 Pharmaceutical particulars
8.1 Incompatibilities
Not applicable
8.2 Shelf-life
Refer on pack
8.3 Packaging information
8.4 Storage and handing instructions
Store protected from light & moisture at a temperature not exceeding 30°C.
Keep out of reach of children.
9.0 Patient Counselling Information
- Instruct patients to take ZOSA DSR Capsules exactly as prescribed by doctor. Do not change the dose or stop therapy without consulting to your doctor.
- Instruct patients to swallow ZOSA DSR Capsules whole with water on empty stomach, preferably in the morning or at least 1 hour before meal. The capsules should not be opened, chewed or crushed.
- If you miss a dose, take it as soon as possible. If it is almost time for your next dose, do not take the missed dose. Take the next dose at your regular time. Do not take 2 capsules/doses at the same time to make up for the missed dose.
- Pregnant women can use this medicine only if essential and in consultation with their doctor.
- Advise nursing mothers to avoid use of this medicine during lactation or not to breastfeed their infants while on drug therapy.
- This medicine is not recommended for use in children.
- Instruct patients not to share this medication with other people even though symptoms are similar. It may harm them.
- Tell your doctor about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements. ZOSA DSR Capsules and certain other medicines can interact with each other causing serious side effects.
12.0 Date of revision
17.09.2022
About Leaflet
Read all of this leaflet carefully before you start taking this medicine
- Keep this leaflet. You may need to read it again.
- If you have any further questions, ask your doctor, health care provider or pharmacist.
- This medicine has been prescribed for you only. Do not pass it on to others. It may harm them, even if their symptoms are the same as yours.
- If any of the side effects gets serious, or if you notice any side effects not listed in this leaflet, please tell your doctor, health care provider or pharmacist.
In this leaflet:
1. What Zosa DSR is and what it is used for
2. Before you take Zosa DSR
3. How to take Zosa DSR
4. Possible Side Effects
5. How to store Zosa DSR
6. Further information
1. What Zosa DSR is and what it is used for
Esomeprazole contains a medicine called esomeprazole. This belongs to a group of medicines called ’proton pump inhibitors’. They work by reducing the amount of acid that your stomach produces. Domperidone contain domperidone as the active ingredient, which belongs to a group of medicines called 'dopamine antagonists'.
How Zosa DSR works
Esomeprazole is used to treat the following conditions: Adults and adolescents aged 12 years and above
- ‘Gastroesophageal reflux disease’ (GORD). This is where acid from the stomach escapes into the gullet (the tube which connects your throat to your stomach) causing pain, inflammation and heartburn.
- Ulcers in the stomach or upper part of the gut (intestine) that are infected with bacteria called ‘Helicobacter pylori’. If you have this condition, your doctor may also prescribe antibiotics to treat the infection and allow the ulcer to heal.
- Domperidone works by helping to move food faster through your food pipe (esophagus), stomach and gut. This is so that it does not stay in the same place for too long. It also helps stop food flowing the wrong way back up your food pipe. This medicine is for use in adults and children aged 16 years and over.
2. Before you take Zosa DSR
Do not take Esomeprazole & Domperidone:
If you have an allergy to:
- Any medicines containing esomeprazole, amoxicillin or clarithromycin
- Any ingredients listed at the end of the leaflet
- Any other similar medicines such as proton pump inhibitors, penicillins, cephalosporins or macrolide antibiotics. Some of the symptoms of an allergic reaction may include shortness of breath, wheezing or difficulty breathing; swelling of the face, lips, tongue or other parts of the body; rash, itching or hives on the skin.
Do not take Esomeprazole & Domperidone if you are taking any of the following medicines:
- Cisapride
- Pimozide
- Ergotamine
- Dihydroergotamine
- Astemizole
- Terfenadine
- Cilostazol
- Atazanavir
- Colchicine
- Simvastatin
- Lovastatin.
Taking Zosa DSR and Other medicines
Do not take Esomeprazole & Domperidone if you are taking the following medicines:
- Cisapride, a medicine used to treat heartburn
- Pimozide, a medicine used to treat mood disorders
- Ergotamine or dihydroergotamine, medicines used to treat migraine headaches
- Astemizole or terfenadine, medicines used to treat hay fever and allergies
- Atazanavir, a medicine used to treat Human Immunodeficiency Virus (HIV)
- Cilostazol, a medicine used to treat intermittent claudication.
- Colchicine, a medicine used to treat gout
- Lovastatin or simvastatin, medicines used to treat high cholesterol
In particular, tell your doctor if you are taking any of the following:
- Ketoconazole tablets or liquid for fungal infections.
- Antibiotics for infections (such as erythromycin)
Pregnancy and breast-feeding
Before taking Esomeprazole & Domperidone, tell your doctor if you are pregnant or trying to get pregnant. Ask your doctor or pharmacist for advice before taking any medicine. Your doctor will decide whether you can take Esomeprazole & Domperidone during this time. It is not known if passes into breast milk. Therefore, you should not take Esomeprazole & Domperidone if you are breastfeeding. If you have not told your doctor about any of the above, tell them before you start taking Esomeprazole & Domperidone.
3. How to take Zosa DSR
Follow all directions given to you by your doctor or pharmacist carefully.
They may differ from the information contained in this leaflet.
If you do not understand the instructions on the box, ask your doctor or pharmacist for help.
- If your doctor has found that your food pipe (gullet) has been slightly damaged, the recommended dose is one Capsule of Esomeprazole & Domperidone once a day for 4 weeks. Your doctor may tell you to take the same dose for a further 4 weeks if your gullet has not yet healed.
- The recommended dose once the gullet has healed is one Capsule once a day.
It is gastro-resistant capsules are not recommended for children less than 12 years old.
- If you are taking this medicine for a long time, your doctor will want to monitor you (particularly if you are taking it for more than a year).
- If your doctor has told you to take this medicine as and when you need it, tell your doctor if your symptoms change.
You can take your capsules at any time of the day.
- You can take your capsules with food or on an empty stomach.
- Swallow your capsules whole with a drink of water. Do not chew or crush the capsules. This is because the capsules contain coated pellets which stop the medicine from being broken down by the acid in your stomach. It is important not to damage the pellets.
4. Possible side effects
Tell your doctor or pharmacist as soon as possible if you do not feel well while you are taking Esomeprazole & Domperidone.
Esomeprazole & Domperidone helps most people with peptic ulcer and Helicobacter pylori infection, but it may have unwanted side effects in a few people. All medicines can have side effects. Sometimes they are serious, most of the time they are not. You may need medical treatment if you get some of the side effects.
Ask your doctor or pharmacist to answer any questions you may have.
Tell your doctor if you notice any of the following and they worry you:
- Diarrhoea, nausea or vomiting
- Headache
- Dizziness
- Constipation
- Loss of appetite
- Stomach pain
- Wind
- Dryness of the mouth or other body cavities
- Soreness of mouth or tongue
- "pins and needles"
- Metallic taste or other change in taste or smell.
- These side effects are usually mild.
Tell your doctor immediately or go to Accident and Emergency at your nearest hospital if you notice the following:
- Muscle pain or weakness
- Increase in breast size (males)
- Fever
- Changes in sleep patterns
- Mood changes, hallucinations, confusion or depression
- Change in sexual function
- Blurred vision, hearing disturbances
- Hair loss
- bleeding or bruising more easily than normal
- Signs of frequent infections such as fever, severe chills, sore throat or mouth ulcers
- Skin rash
- Blood in urine
- Tremor
- Convulsions or fits
- Overgrowth of yeast infections (thrush).
Do not take any medicine to stop the diarrhoea unless advised by your doctor. These are very serious side effects. If you have them, you may have had a serious reaction to one of the medicines in Esomeprazole & Domperidone. You may need urgent medical attention or hospitalization.
Tell your doctor if you notice anything else that is making you feel unwell. Some people may get other side effects while taking Esomeprazole & Domperidone. Do not be alarmed by this list of possible side effects. You may not experience any of them.
Reporting of side effects
If you get any side effects, talk to your doctor, pharmacist or nurse. This includes any possible side effects not listed in this leaflet. You can also report side effects directly: Website: www.zuventus.com and click the tab “Safety Reporting” located on the top of the home page.
By reporting side effects, you can help provide more information on the safety of this medicine.
You can also report the side effect with the help of your treating physician.
5. How to store Zosa DSR
Keep out of the reach and sight of children.
Do not store above 30°C.
Do not use Zosa DSR after the expiry date which is stated on the label/carton/bottle after EXP. The expiry date refers to the last day of the month.
Do not use Zosa DSR If you notice the visible signs of deterioration.
Medicines should not be disposed of via wastewater or household waste. Ask your pharmacist how to dispose of medicines no longer required. These measures will help to protect the environment.
6. Further information
What Zosa DSR contains
Each hard gelatin capsule contains:
Esomeprazole Magnesium (Trihydrate) IP
equivalent to Esomeprazole …………………. 40 mg
(as gastro-resistant pellets)
Domperidone IP …………………………………… 30 mg
(as prolonged release pellets)
Marketing Authorisation Holder
Zuventus Healthcare Limited
Zuventus House Plot No. Y2, CTS No: 358/A2,
near Nahur Railway Station, off Raycon IT Park Road,
Nahur West, Mumbai, Maharashtra 400078
Date of revision
This leaflet was last revised in 09/2024.